ABC mukowiscydozy – fakty na temat choroby
Poniżej przedstawiamy ciągle rozbudowywaną bazę informacji na temat mukowiscydozy. Są to najczęściej wyszukiwane informacje, pytania czytelników i chorych na mukowiscydozę.
1. Podstawowe informacje o mukowiscydozie (13)
Mukowiscydoza, inaczej zwłóknienie torbielowate (w skrócie CF od angielskiej i łacińskiej nazwy Cystic Fibrosis) jest zaburzeniem genetycznym, głównie płuc, ale także trzustki , wątroby , nerek i jelit. Jest to choroba genetyczna.
Ocenia się, że obecnie na świecie żyje ponad 100 tysięcy osób z mukowiscydozą, głównie w USA, Kanadzie, Australii i Europie.
Mukowiscydoza to choroba genetyczna. Nosiciele genu odpowiedzialnego za wystąpienie mukowiscydozy stanowią około 5% ludzi rasy białej. Para dwóch nosicieli wadliwego genu ma 25% szans na chorego potomka.
Mukowiscydoza to choroba genetyczna. Nie ma możliwości zarażenia się od chorych na mukowiscydozę, tą chorobą. Nie jest możliwe przeniesienie tej choroby na zdrową osobę (jest to naukowo, fizycznie niemożliwe).
Mukowiscydoza rozwija się jedynie u osób, które otrzymały od obojga rodziców wadliwy gen CFTR.
W Polsce używa się nazwy “mukowiscydoza” lub skrótu “CF”. W starszych publikacjach, zwłaszcza naukowych pojawia się też określenie “zwłóknienie torbielowate”. W języku angielskim używa się nazwy “cystic fibrosis” lub “CF”.
Mukowiscydoza to choroba śmiertelna. Długość przeżycia zależy od bardzo wielu czynników.
Połowa chorych w Polsce obecnie dożywa jedynie około 24 lat [1]. Co roku umierają w Polsce dzieci, młodzież i młode osoby z mukowiscydozą.
Średnia długość życia chorego na mukowiscydozę na świecie jest różna w zależności od kraju. W Kanadzie w 2013 r. średnia długość życia u osób z mukowiscydozą wynosiła 50,9 lat, w USA 40,6 lat.
[1] Dane z 2014 roku.
Nie ma skutecznej metody leczenia mukowiscydozy i zapobiegania progresji tej choroby. Mukowiscydoza jest chorobą śmiertelną. Przyjmuje się, że średnia długość życia chorego na mukowiscydozę w Polsce obecnie wynosi około 35 lat i ciągle, stopniowo od wielu lat się wydłuża. Na długość życia chorego z CF ma wpływ wiele czynników, w tym sposób leczenia i fizjoterapii, choroby towarzyszące, liczba i stopień zaostrzeń.
Trwają liczne międzynarodowe badania naukowe nad stworzeniem skutecznych leków i terapii mukowiscydozy oraz chorób towarzyszących w CF. Więcej o nowych terapiach i lekach przeczytasz w dziale aktualności: Nauka
Choroby rzadkie to bardzo rzadko występujące schorzenia uwarunkowane najczęściej genetycznie, o przewlekłym i często ciężkim przebiegu, w około połowie ujawniają się w wieku dziecięcym. Ze względu na rzadkość występowania, trudności w rozpoznawaniu i brak świadomości społecznej, wiedza o tych chorobach była dotychczas niewielka.
Dotychczas wykryto ponad sześć tysięcy rzadkich chorób.
Mukowiscydoza jest najczęściej występującą chorobą rzadką w Polsce oraz na świecie.
Nie! Mukowiscydoza nie wiąże się z niepełnosprawnością intelektualną chorego. Mutacja genu CFTR występująca w mukowiscydozie, ani choroby towarzyszące (współistniejące typu cukrzyca) nie wpływają na sprawność intelektualną.
Atypowa mukowiscydoza, zwana też nietypową lub nieklasyczną mukowiscydozą, jest łagodniejszą postacią “mukowiscydozy”.
Zamiast klasycznych objawów, osoby z atypową mukowiscydozą mogą mieć jedynie łagodne dysfunkcje w jednym układzie narządów. Stężenie chlorków w pocie często nie przekracza poziomu 60mmol/l, a u wielu chorych z atypową mukowiscydozą nie przekracza nawet 30mmol/l (czyli poziom chloru w pocie jest całkowicie w normie).
Atypowa mukowiscydoza jest bardzo zróżnicowanym zaburzeniem wpływającym na różne układy narządów w różnym stopniu. Objawy, których doświadczają pacjenci, mogą również ulegać zmianom w czasie; jednak niektóre kliniczne objawy przedmiotowe i podmiotowe dotyczące układu oddechowego, żołądkowo-jelitowego, hormonalnego i metabolicznego oraz układu moczowo-płciowego powinny ostrzegać lekarzy o możliwości wystąpienia mukowiscydozy. Pacjenci z atypowym mukowiscydozą często mają mniej hospitalizacji w okresie dzieciństwa niż ci z klasyczną mukowiscydozą, a zaburzenie może pozostać niezdiagnozowane przez wiele lat (mowa o osobach nie objętych badaniami przesiewowymi, w Polsce przed 2009 rokiem). Najstarszą opisaną w literaturze naukowej, osobą z atypową mukowiscydozą, była kobieta w wieku 70 lat w momencie postawienia diagnozy.
Warunkiem rozpoznania atypowej mukowiscydozy jest zidentyfikowanie patogennych mutacji CFTR w obu allelach.
Ocenia się, że w zależności od kraju, liczba chorych z atypową mukowiscydozą w ogólnej populacji chorych może wynosić w granicach 1-9%.
Osoby z atypową mukowiscydozą mają zazwyczaj 1 ciężką mutację klasy I lub II i 1 łagodniejszą mutację lub nieprawidłowość powtórzeń trinukleotydów na ich innym genie CFTR.
W atypowej mukowiscydozie, objawy ze strony układu oddechowego są często łagodniejsze i mogą pojawić się dopiero w wieku dorosłym i przypominać objawy astmy lub przewlekłej obturacyjnej choroby płuc (POChP, ang. COPD).
W atypowej mukowiscydozie efekty żołądkowo-jelitowe mogą być bardzo subtelne i obejmować głównie przewlekłe zaparcia lub biegunki. U niektórych pacjentów może występować przewlekłe lub nawracające zapalenie trzustki.
CFTR z języka ang. cystic fibrosis transmembrane conductance to białko tworzące kanał chlorkowy w błonie komórkowej, kodowane przez gen CFTR. Jego nieprawidłowa forma (spowodowana mutacjami genu CFTR) wywołuje chorobę genetyczną zwaną mukowiscydozą.
CFTR-RD z angielskiego CFTR-related disorders, można zdefiniować jako “jednostkę kliniczną powiązaną z dysfunkcją CFTR, która nie spełnia kryteriów diagnostycznych dla mukowiscydozy” ani dla postaci typowej, klasycznej ani dla mukowiscydozy atypowej.
CFTR-RD charakteryzuje się występowaniem pojedynczych objawów powiązanych z występowaniem najczęściej jednej mutacji CFTR.
W CFTR-RD diagnozuje się:
- CBAVD, czyli niedrożność i aplazja nasieniowodów z dysfunkcją CFTR,
- ostre nawracające lub przewlekłe zapalenie trzustki z dysfunkcją CFTR
- rozstrzenie oskrzeli z dysfunkcją CFTR.
Mukowiscydoza to najczęstsza “choroba rzadka” w Polsce.
Według różnych źródeł ocenia się, że w Polsce żyje ponad 1800-2000 chorych na mukowiscydozę. Niektóre analizy wskazują, że liczba ta może być większa i wynosić nawet 3 tysiące chorych.
Dlaczego nie znamy dokładnej liczby chorych? Bo w Polsce od wielu lat nie prowadzono rejestru chorych na mukowiscydozę (do 2012 był prowadzony rejestr chorych, ale nie był kompletny, dane pochodziły tylko z części ośrodków leczących chorych na mukowiscydozę). Zmienić ma się to dopiero w tym roku bo Polska dołączyła do Europejskiego Rejestru CF (European Cystic Fibrosis Society Patient Registry) i trwają przygotowania do wdrożenia systemu przekazywania informacji w placówkach medycznych.
2. Badania genetyczne w kierunku mukowiscydozy, badania przesiewowe, rozpoznanie mukowiscydozy (14)
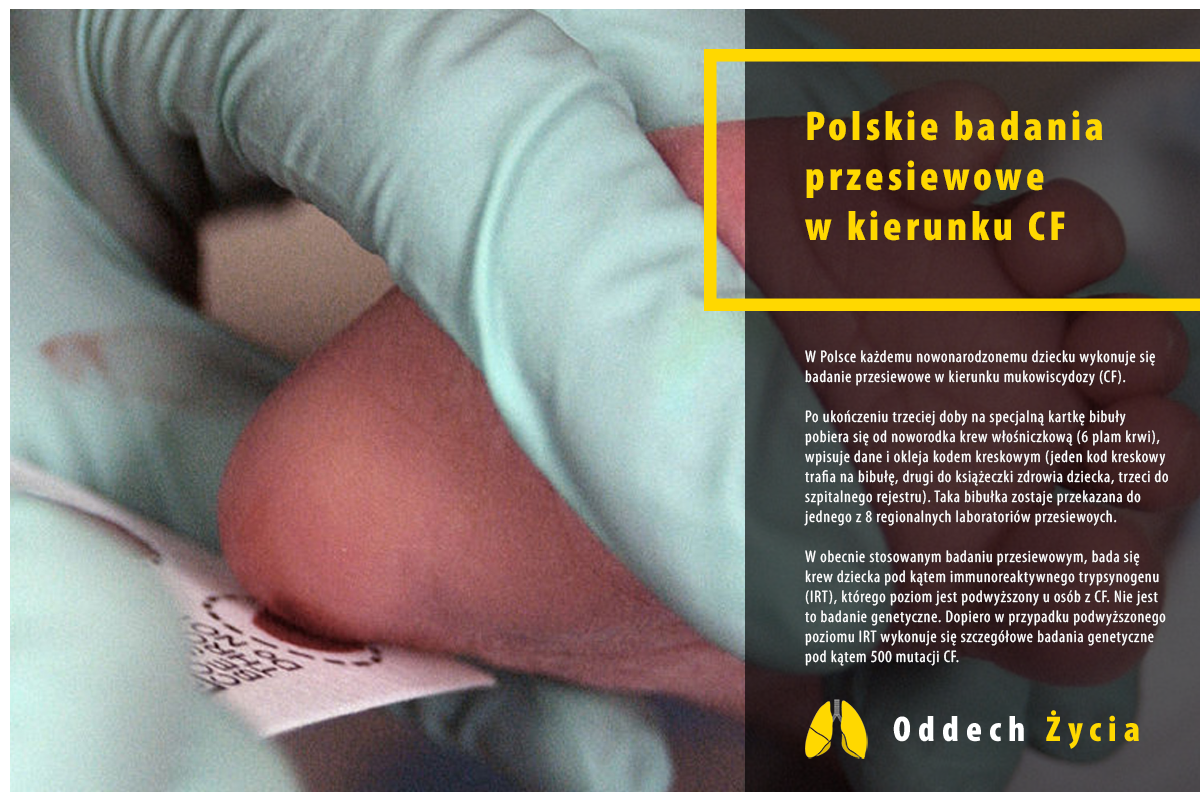
Badania przesiewowe w kierunku mukowiscydozy wykonuje się każdemu nowo narodzonemu dziecku w polskich szpitalach. Nie są 100% skuteczne, ocenia się że wykrywają 99,5% przypadków CF.
Polegają na pobraniu krwi noworodka i zbadaniu poziomu trypsynogenu.
Jeżeli Ty lub Twój partner/ka boicie się ryzyka mukowiscydozy bo w rodzinie występowały już przypadki tej choroby możecie skorzystać z konsultacji Poradni Genetycznej. Specjalista może zlecić badania genetyczne w kierunku nosicielstwa zmutowanego genu CFTR. Skierowanie do poradni może wystawić zarówno lekarz POZ jak i inny lekarz specjalista (bez skierowania wizyta konsultacyjna będzie odpłatna).
Większość ludzi w Polsce i na świecie nie wie o nosicielstwie wadliwego genu CFTR odpowiedzialnego za mukowiscydozę. Jeżeli masz obawy, to skorzystaj z konsultacji Poradni Genetycznej, która może zlecić badania genetyczne lub test potowy (badanie diagnostyczne potwierdzające mukowiscydozę).
W przypadku potomstwa dwóch nosicieli zmutowanego genu CFTR istnieje jedna na cztery szanse na przeniesienie dwóch zmutowanych genów, po jednym od każdego rodzica (dziecko z dwoma zmutowanymi genami CFTR jest chore na mukowiscydozę). Dlatego w rodzinach, w których rodzice są nosicielami genu CFTR zawsze występuje ryzyko wystąpienia mukowiscydozy.
Lista poradni genetycznych w Polsce wykonujących konsultacje i badania w kierunku mukowiscydozy oraz innych chorób genetycznych.
Poradnia Genetyczna Zakładu Genetyki Klinicznej UM
ul. Waszyngtona 17
15-274 Białystok
tel.: (0-85) 745-08-01
NZOZ Podlaskie Centrum Genetyki Klinicznej GENETICS
ul. Parkowa 14/84
15-224 Białystok
tel.: ( 0-85) 742-64-26
Rejestracja telefoniczna
Poradnia Genetyczna, SPZOZ Wojewódzki Szpital Zespolony
im. J. Śniadeckiego
ul. Warszawska 15
15-062 Białystok
tel.: (0-85) 748-87-00 wew. 745
Poradnia Genetyczna Katedry i Zakładu Genetyki Klinicznej AM
ul. Marii Skłodowskiej-Curie 9
85-094 Bydgoszcz
tel.:(0-52) 585-43-67
Rejestracja telefoniczna
Poradnia Genetyczna
Katedry i Zakładu Biologii i Genetyki UM
ul. Dębinki 7
80-210 Gdańsk
tel.: (0-58) 349-34-68
Rejestracja telefoniczna
Poradnia Genetyczna
Górnośląskie Centrum Zdrowia Dziecka SUM
ul. Medyków 16
40-752 Katowice
tel.:(0-32) 207-16-41
Rejestracja telefoniczna
Poradnia Genetyczna Zakładu Genetyki Klinicznej Polsko-Amerykański Instytut Pediatrii UJ
ul. Wielicka 265
30-683 Kraków
tel.: (0-12) 658-20-11
NZOZ Poradnia Genetyczna Kostyk i Kruczek Spółka Partnerska Lekarzy
ul. Wieniawskiego 64
31-436 Kraków
tel.: (0-12) 418-81-60, fax: (0-12) 418-81-61
www.poradniagenetyczna.pl
Poradnia Genetyczna Zakładu Genetyki Medycznej UM SP Szpital Wojewódzki im. Jana Bożego
ul. Piłsudskiego 11
20-093 Lublin
tel.: (0-81) 532-05-35
Rejestracja telefoniczna
Poradnia Genetyczna Katedry Genetyki Medycznej i Zakładu Genetyki Medycznej Instytut Endokrynologii UM
ul. Sterlinga 5
91-425 Łódź
tel.: (0-42) 632-27-42
Rejestracja telefoniczna
Poradnia Genetyczna Centrum Zdrowia Matki Polki
ul. Rzgowska 281/289
93-338 Łódź
tel.: (0-42) 271-11-31
Rejestracja telefoniczna i osobista
Poradnia Genetyczna NZOZ “GENOS”
ul. Piotrkowska 204/210
90-338 Łódź
tel. (0-42) 611-63-11
Poradnia Genetyczna, Samodzielny Zespół Publicznych Zakładów Opieki Zdrowotnej im. “Dzieci Warszawy” w Dziekanowie Leśnym
ul. M.Konopnickiej 65
05-092 Łomianki
tel.: (0-22) 765-71-73
Rejestracja telefoniczna
Poradnia Genetyczna Woj. Specjalistycznego Szpitala Dziecięcego
ul. Żołnierska 18
10-560 Olsztyn
tel.: (0-81) 539-32-00,539-32-01
Rejestracja telefoniczna
Godziny przyjęć: wtorki 9:00 – 15:00
Poradnia Genetyczna
Centrum Genetyki Medycznej NZOZ
ul. Grudzieniec 4
60-823 Poznań
tel.: (0-61) 852-73-32, 848-40-38
Rejestracja telefoniczna
Poradnia Genetyczna
SP ZOZ nr 1
ul. Hetmańska 21
35-045 Rzeszów
tel.: (0-17) 853-52-81, wew. 352
Rejestracja telefoniczna
Poradnia Genetyczna
SPSK Nr 2
ul. Połabska 4
70-115 Szczecin
tel.: (0-91) 466-15-65
Rejestracja telefoniczna
Godziny przyjęć: poniedziałek-piątek 8:00 – 12:00
Poradnia Genetyczna, Zakładu Genetyki Medycznej Instytut-Pomnik Centrum Zdrowia Dziecka
Al. Dzieci Polskich 20
04-736 Warszawa-Międzylesie
tel.: (0-22) 327-71-38
Rejestracja telefoniczna
Poradnia Genetyczna, Zakładu Genetyki Medycznej Instytut Matki i Dziecka
ul. Kasprzaka 17a6
01-211 Warszawa
tel.: (0-22) 327-71-38
Rejestracja telefoniczna
Poradnia Genetyczna, Zakładu Genetyki, Instytut Psychiatrii i Neurologii
Al. Sobieskiego 1/9
02-957 Warszawa
tel.: (0-22) 458-26-10, 458-28-56
Rejestracja telefoniczna
Samodzielny Publiczny Dziecięcy Szpital Kliniczny
Poliklinika II
Poradnia genetyczna dla dzieci
ul. Działdowska 1
01-184 Warszawa
tel. (0-22) 45-232-67
Rejestracja telefoniczna
Poradnia Genetyczna
Zakładu Genetyki Medycznej
Katedra i Zakład Patofizjologii AM
ul. Marcinkowskiego 1
50-368 Wrocław
tel.: (0-71) 784-12-55, 784-27-25, 327-09-74
Godziny przyjęć: poniedziałek 11:00 – 18:00
wtorek, czwartek, piątek 9:00 – 14:00
Jeżeli nie znalazłeś poradni w swojej okolicy lub znasz poradnię specjalizującą się poradnictwem i diagnozowaniem w kierunku mukowiscydozy, której nie ma na powyższej liście, to skontaktuj się z nami pod adresem [email protected]
Obecnie znamy ponad 2000 różnych mutacji genu CFTR. Nie wszystkie mutacje CFTR wywołują objawy mukowiscydozy, prowadzą do rozwoju mukowiscydozy, niektóre mogą nieść ryzyko wystąpienia objawów mukowiscydozy (CFSPID), a niektóre mogą nie wywoływać żadnych objawów mukowiscydozy.
CFSPID
W trakcie diagnostyki (zwykle podczas badań przesiewowych) stwierdza się czasem obecność mutacji o nieznanych konsekwencjach klinicznych CFSPID. Część dzieci z CFSPID do końca życia nie będzie chorować na mukowiscydozę i choroby towarzyszące mukowiscydozie. U części dzieci z CFSPID może rozwinąć się mukowiscydoza oraz choroby towarzyszące (zależne od mutacji CFTR). W takim przypadku mówimy o wykryciu jednej lub dwóch mutacji o nieznanym rokowaniu.
Atypowa mukowiscydoza
U nie wielkiej części osób z dwiema mutacjami może rozwinąć się mukowiscydoza atypowa. Osoby z atypową mukowiscydozą mają zazwyczaj 1 ciężką mutację klasy I lub II i 1 łagodniejszą mutację lub nieprawidłowość powtórzeń trinukleotydów na ich innym genie CFTR.
Klasyczna mukowiscydoza
W trakcie diagnostyki wykrywa się dwie mutacje chorobotwórcze, patogenne, wywołujące mukowiscydozę.
Niepatogenne mutacje
W trakcie diagnostyki mogą zostać odkryte tez mutację nie wywołujące objawów mukowiscydozy. U takich osób nie rozwinie się mukowiscydoza. Mimo mutacji będą zdrowe.
CFTR-RD
CFTR-RD charakteryzuje się występowaniem pojedynczych objawów mukowiscydozy powiązanych z występowaniem najczęściej jednej mutacji CFTR. Ale nie mówimy tutaj o mukowiscydozie, a chorobach zależnych od mutacji CFTR.
Rodzice w Polsce są informowani o rodzaju mutacji (list z wynikami laboratoryjnymi, genetycznymi zawiera informację, czy mutacja jest patogenna/chorobotwórcza lub o nieznanych konsekwencjach klinicznych CFSPID).
Badanie chlorków w pocie (test potowy) jest istotnym badaniem w diagnostyce mukowiscydozy.
Podwyższony, wysoki poziom chlorków w pocie może występować nie tylko w mukowiscydozie. Dlatego w tej chorobie test potowy jest częścią szerszych badań diagnostycznych, najczęściej połączonych z wykonaniem badań genetycznych. Od 2009 jest zwykle konsekwencją pozytywnego wyniku badań przesiewowych w kierunku mukowiscydozy. Wykonuje się go też u dzieci i osób starszych, u których wystąpiły objawy mukowiscydozy, w celu potwierdzenia diagnozy.
Oprócz mukowiscydozy, podwyższony poziom chlorków w pocie może występować u osób z:
- niedoczynnością kory nadnerczy,
- anoreksją,
- dysautonomią (dysfunkcja autonomiczna, neuropatia autonomiczna),
- dysplazją ektodermalna,
- egzemą (eczema),
- fukozydozą
- niedoborem dehydrogenazy glukozo-6-fosforanowej (fawizm),
- chorobą von Gierkego,
- niedoczynnością przytarczyc,
- niedoczynnością tarczycy,
- niedożywieniem z różnych przyczyn,
- moczówką prostą,
- postępującą cholestazą G6PD.
Test potowy służy do pomiaru ilość chlorków w pocie. Osoby dotknięte mukowiscydozą mogą mieć od dwóch do pięciu razy większą ilość chlorków w pocie.
Referencyjną metodą pomiaru ilości chlorków w pocie w mukowiscydozie jest klasyczny test potowy – ilościowa jonoforeza pilokarpinowa. Alternatywną dla jonoforezy pilokarpinowej jest test konduktometryczny, który pozwala uzyskać wynik w krótszym czasie, również u bardzo małych dzieci.
Test chlorków w pocie (test potowy) jest istotnym badaniem w diagnostyce mukowiscydozy. Rzadko, ale jednak możliwe są fałszywie ujemne, negatywne wyniki testu potowego w mukowiscydozie. Czyli mimo mukowiscydozy, wyniki będą “w normie”.
Takie wyniki możemy otrzymać między innymi w przypadku:
- niektórych mutacji patogennych (odpowiedzialnych za mukowiscydozę) oraz mutacji o niejasnych konsekwencjach klinicznych, np. 3849+10kbC->T,
- obrzęków obwodowych, zwłaszcza u niemowląt z hipoproteinemią,
- dużego niedożywienia,
- błędów technicznych sprzętu lub nieprawidłowego sposobu przeprowadzenia badania,
- stosowania mineralokortykosteroidów.
Zobacz też “Przyczyny dodatniego testu potowego (podwyższonych chlorków) mimo braku mukowiscydozy“.
W Polsce test potowy można wykonać już u małych dzieci – noworodków urodzonych w terminie, powyżej 10 dnia życia i masie ciała powyżej 2 kg (są odstępstwa od tej reguły u dzieci z podejrzeniem mukowiscydozy w wyniku niedrożności smółkowej).
Test potowy w diagnostyce mukowiscydozy nie wymaga specjalnego przygotowania, dziecko nie musi być na czczo. Przed wykonaniem tego testu dziecko może jeść, pić i ćwiczyć, jak zwykle oraz kontynuować przyjmowanie jakichkolwiek leków.
Najczęściej zaleca się, by nie kremować rąk i nóżek u noworodka 24h przed wykonywaniem badania.
Badanie jest czasochłonne. Warto zabrać zabawki, które zajmą uwagę dziecka przez czas trwania badania.
Górną granicą ilości chlorków w pocie to wynik 29 mmol/l, a wynikiem prawidłowym każdy od 0 do 29 mmol/l włącznie.
Wartości 30-59 mmol/l wskazują na niejednoznaczne wyniki i na podstawie jedynie testu potowego nie można potwierdzić lub wykluczyć mukowiscydozy. Najczęściej w takim przypadku wykonuje się rozszerzone badania genetyczne.
Wartości powyżej 60 mmol/l mogą wskazywać na mukowiscydozę lub fałszywie dodatni wynik testu potowego.
Badanie przeznabłonkowej różnicy potencjałów w błonie śluzowej nosa (w języku angielskim Nasal Transepithelial potential difference lub NPD) to użyteczne narzędzie diagnostyczne w przypadku niejednoznacznych wyników badań diagnostycznych w kierunku mukowiscydozy.
Badanie polega na pomiarze różnicy potencjałów, tj. ładunku na powierzchni nabłonka dróg oddechowych w nosie w porównaniu z płynem śródmiąższowym. Osoby z mukowiscydozą mają znacznie bardziej ujemną powierzchnię błony śluzowej nosa niż normalnie, ze względu na zwiększone wchłanianie sodu.
Badanie NPD wykonuje się in vivo, czyli na pacjencie, w odróżnieniu od badania ICM. Badanie NPD jest bezpieczne i nieinwazyjne dla pacjenta.
Badanie ICM z ang. Intestinal current measurement.
Podobnie jak badanie przeznabłonkowej różnicy potencjałów w błonie śluzowej nosa (NPD), ICM może być przydatne w diagnozowanie mukowiscydozy w przypadku niejednoznacznych wyników innych badań diagnostycznych. W odróżnieniu od NPD, badanie ICM jest dość łatwe do wykonania w każdym wieku, także u niemowląt.
Badanie polega na pobraniu komórek błony śluzowej odbytu lub jelita czczego. Pobrane komórki już w warunkach laboratoryjnych umieszcza się komorze Ussinga, która służy do pomiaru epitelialnego transportu jonów.
3. Objawy mukowiscydozy (6)
Trudno jednoznacznie odpowiedzieć na to pytanie. Mukowiscydoza rozwija się w różnym tempie. Zależy to od wielu czynników, w tym mutacji genetycznych powodujących mukowiscydozę, postępowania dietetycznego, liczby zaostrzeń płucnych i wielu innych czynników. Zwykle pierwsze objawy mukowiscydozy ujawniają się już w wieku niemowlęcym.
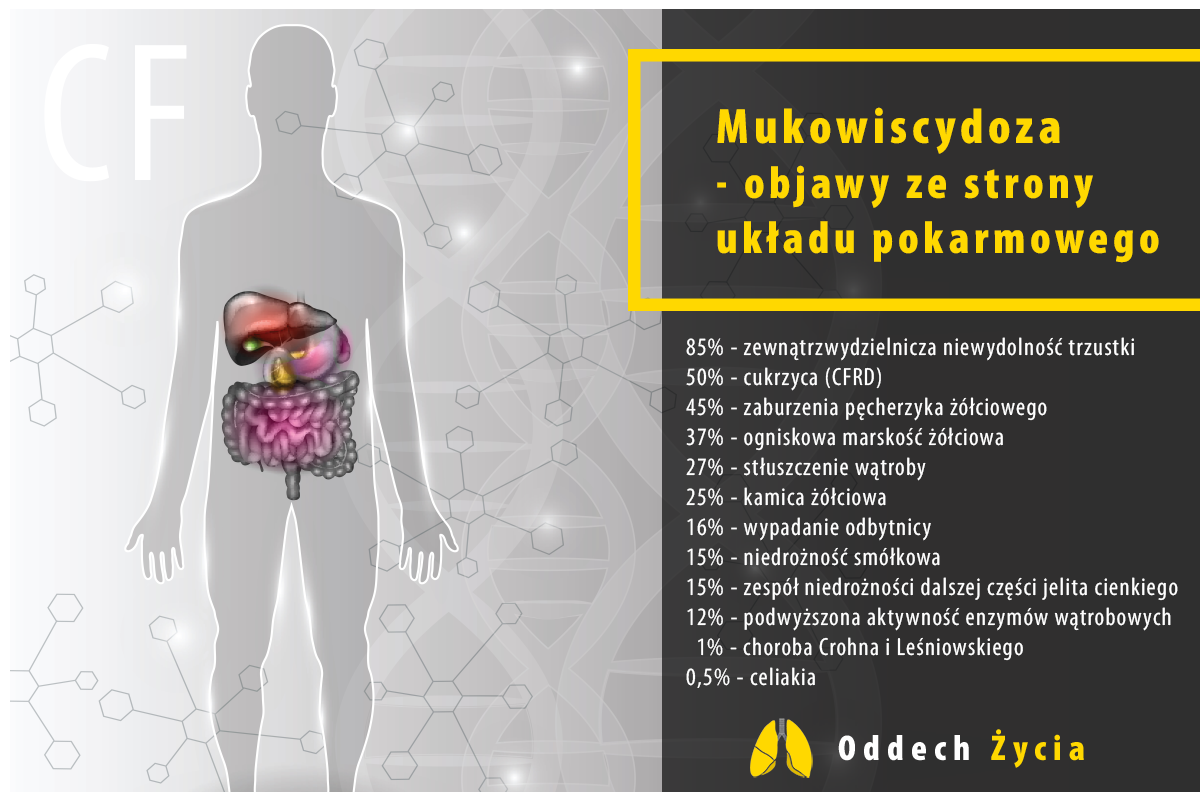
Do najczęstszych objawów mukowiscydozy i chorób towarzyszących związanych z układem pokarmowym należą:
- zewnątrzwydzielnicza niewydolność trzustki (zaburzenia odżywiania, niedowaga, tłuste biegunki, wzdęcia i bóle brzucha)
- cukrzyca (CFRD)
- zaburzenia pęcherzyka żółciowego
- ogniskowa marskość żółciowa
- stłuszczenie wątroby
- kamica żółciowa
- wypadanie odbytnicy
- niedrożność smółkowa
- zespół niedrożności dalszej części jelita cienkiego
- podwyższona aktywność enzymów wątrobowych
- choroba Crohna i Leśniowskiego
- celiakia
![Objawy mukowiscydozy ze strony układu pokarmowego]()
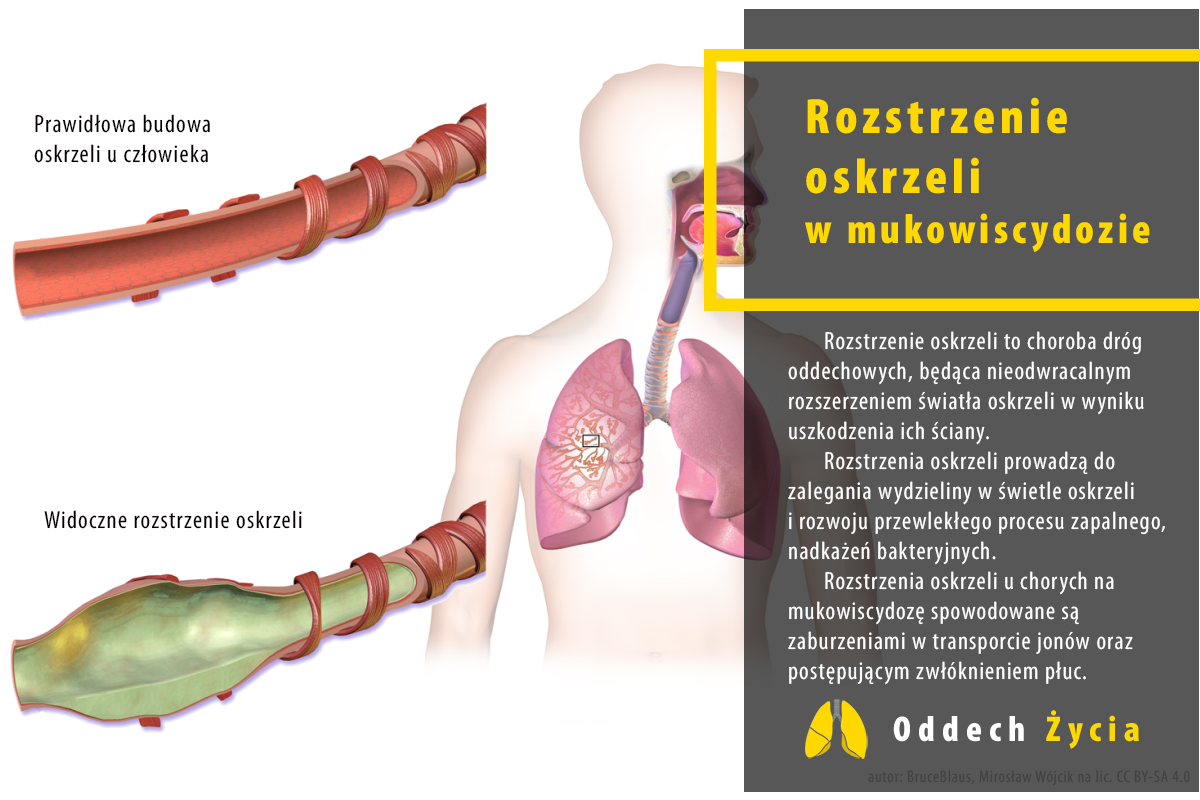
Rozstrzenie oskrzeli to choroba dróg oddechowych, będąca nieodwracalnym rozszerzeniem światła oskrzeli w wyniku uszkodzenia ich ściany. Medyczna nazwa tej jednostki chorobowej “Bronchiectasis” pochodzi od greckich słów “bronckos” (drogi oddechowe) i “ektasis” (poszerzanie).
Rozstrzenie oskrzeli może być wrodzone lub nabyte. W mukowiscydozie, pierwotnej dyskinezie rzęsek (PCD) mówimy o etiologii wrodzonej.
Jakie są objawy rozstrzenia oskrzeli?
- uporczywy kaszel
- odkrztuszanie dużej ilości wydzieliny, zazwyczaj ropnej
- krwioplucie
- cuchnący oddech
- stany podgorączkowe
- duszność początkowo wysiłkowa, następnie spoczynkowa
Choroba rozwija się powoli, latami, z okresami zaostrzeń, z nawracającymi bakteryjnymi zapaleniami oskrzeli. Stopniowo nasila się kaszel, zwiększa się ilość odpluwanej wydzieliny, pojawia się duszność. W stanach zaawansowanych występują: sinica, palce pałeczkowate, wyniszczenie, niewydolność oddechowa.
Diagnostyka rozstrzeni oskrzeli
Podczas badania fizykalnego – osłuchiwania można stwierdzić rzężenia i szmery oskrzelowe. Badaniem rozstrzygającym o rozpoznaniu rozstrzeni oskrzeli jest tomografia komputerowa wysokiej rozdzielczości (HRCT).
Leczenie rozstrzeni oskrzeli
Nie istnieje leczenie przyczynowe, terapia ma na celu regularne oczyszczanie drzewa oskrzelowego i zapobieganie zakażeniom. Podstawą codziennej terapii jest odpowiednia rehabilitacja oddechowa. W przypadku gdy dojdzie do zakażenia stosuje się antybiotykoterapię.
Niedrożność smółkowa (łac. meconium ileus, ang. MI) to jeden z pierwszych i najcięższych objawów mukowiscydozy. Do niedrożności smółkowej dochodzi u średnio 15% noworodków z mukowiscydozą urodzonych w Polsce. Niedrożność smółkowa nie wskazuje na stopień ciężkości przebiegu mukowiscydozy, ale jest ważną wskazówką dla lekarzy. Szpital powinien poinformować ośrodek prowadzący badania przesiewowe o wystąpieniu niedrożności smółkowej.
Niedrożność smółkowa to stan, w którym treść jelita dziecka (smółka, czyli pierwszy stolec noworodka) jest wyjątkowo lepka i powoduje zablokowanie jelit przy porodzie. W większości przypadków samo jelito jest kompletne i nienaruszone, a tylko wnętrze jelita jest zablokowane. Powodem zbyt gęstej smółki są zaburzenia spowodowane mutacjami CFTR.
Objawy niedrożności smółkowej
- brak pierwszego stolca (smółki) w ciągu pierwszych 24-48 godzin życia,
- zielone wymioty (zwane również żółciowymi, ponieważ zawierają żółć),
- obrzęk brzucha wkrótce po urodzeniu.
Dzieci z bardziej złożonymi problemami – takimi perforacje jelita w wyniku niedrożności smółkowej – mogą mieć bardziej poważne objawy, takie jak te:
- bardzo spuchnięty, bolesny brzuch z powodu stanu zapalnego,
- problemy z oddychaniem, ponieważ nacisk brzucha uniemożliwia wypełnienie płuc powietrzem
Diagnozowanie niedrożności smółkowej
W niektórych przypadkach USG prenatalne wykazuje, że jelito dziecka może być zablokowane. Jeśli lekarz podejrzewa, że jelito dziecka jest zablokowane przed porodem, może zlecić dokładniejsze badania oraz ostrzec zespół medyczny zajmujący się dzieckiem tuż po porodzie.
W większości przypadków lekarze podejrzewają niedrożność smółki w oparciu o objawy kliniczne w ciągu pierwszych kilku godzin lub dni po narodzinach. Naciskając na brzuch dziecka, lekarz może wyczuć pętlę jelita cienkiego (jelito kręte) wypełnioną smółką.
W przypadku podejrzenia niedrożności smółkowej u noworodka zwykle zleca się wykonanie badania RTG. Na zdjęciu rentgenowskim widać powiększone pętle jelita cienkiego.
Leczenie niedrożności smółkowej
U około 60% niemowląt z niedrożnością smółkową możliwe jest rozpuszczenie lepkiej smółki w szpitalu, za pomocą lewatywy.
U niektórych niemowląt mogą wystąpić powikłania, co oznacza, że konieczna jest operacja.
Chirurg podczas operacji przez nacięcie koło pępka, usuwa odcinek jelita, który jest zablokowany i każdy fragment jelita, który został uszkodzony przez zastój smółki. Jeśli pozostała część jelita jest wystarczająco zdrowa, chirurg zszywa razem zdrowe końce jelita i zamyka powłokę brzuszną.
Czasami konieczne jest wyprowadzenie jelita na powierzchnię (stomia) jako tymczasowego leczenia przez kilka tygodni. Ma to miejsce w sytuacji, w której pozostawiona część jelit nie jest w najlepszym stanie. W takim przypadku chirurg przenosi 2 otwarte końce jelita do powierzchni brzucha dziecka i przyszywa do otworów (stomii) wykonanych w ścianie brzucha. Najczęściej kilka tygodni lub miesięcy później podczas kolejnej operacji, zamyka się stomię i zszywa końce jelita.
CFSPID to skrót angielskiej nazwy Cystic Fibrosis Screen Positive, Inconclusive Diagnosis, czyli pozytywny wynik przesiewu noworodkowego, ale niejednoznaczna diagnoza mukowiscydozy. Często przez rodziców diagnoza ta nazywana jest potocznie utajoną mukowiscydozą.
W trakcie diagnostyki stwierdza się obecność mutacji o nieznanych konsekwencjach klinicznych. Często otrzymuje się też graniczne wyniki testu potowego (na granicy).
Część dzieci z CFSPID do końca życia nie będzie chorować na mukowiscydozę i choroby towarzyszące mukowiscydozie. U części dzieci z CFSPID może rozwinąć się mukowiscydoza oraz choroby towarzyszące (zależne od mutacji CFTR). Dlatego dzieci i osoby dorosłe z CFSPID powinny pozostawać pod opieką specjalistyczną w celu obserwacji i diagnostyki, a w razie wystąpienia objawów, wdrożenia odpowiedniego leczenia i fizjoterapii.
Jest to trudna i obciążająca psychicznie diagnoza. Pacjent przez lata nie wie, czy choroba się u niego rozwinie, czy do końca życia będzie żył bez objawów mukowiscydozy.
Zaburzenia węchu (dysosmia) to upośledzenie lub zmiana odczuwania wrażeń węchowych. W wyniku mutacji CFTR i nieprawidłowego działania kanałów chlorkowych dochodzi do zaburzeń klirensu śluzowo-rzęskowego. Ułatwia to kolonizację dróg oddechowych pacjentów z mukowiscydozę przez patogenne drobnoustroje i powoduje częste objawy zapalenia zatok przynosowych, w tym także zaburzenia węchu.
Obecność patogennych bakterii powoduje uwalnianie wiele szkodliwych substancji, które dodatkowo zmniejszają częstotliwość rytmu rzęskowego, a przewlekły stan zapalny powoduje rozrost komórek kubkowych i metaplazję nabłonka. Pod względem makroskopowym czynniki te prowadzą do zatkania zatok szczękowych, powodując zastój śluzu, miejscowy stan zapalny i zaburzenia wymiany gazowej. Powoduje to obrzęk błony śluzowej, zmniejszoną czynność rzęsek i w konsekwencji prowadzi do dalszej kolonizacji bakteryjnej. U niektórych pacjentów z mukowiscydozą dochodzi do rozwoju polipów w nosie, które jeszcze mocniej upośledzają odczuwanie wrażeń węchowych.
Zaburzenia węchu mogą być też skutkiem ubocznym przyjmowania leków, w tym antybiotyków (np. ampicylina, azytromycyna) oraz leków steroidowych.
Co ważne, zmniejszone funkcje węchowe wpływają głównie na progi zapachowe, a nie na identyfikację zapachu. Czyli pacjent prawidłowo rozpoznaje zapachy, ale zmienia się poziom odczuwania wrażeń węchowych. Takie zaburzenie nazywamy hiposmią (osłabieniem powonienia). Rzadziej dochodzi do całkowitej utraty powonienia, zwanej anosmią.
4. Gdzie szukać pomocy w mukowiscydozie? (2)
Specjalistyczne ośrodki w Polsce leczące chorych na mukowiscydozę.
Centrum Leczenia Mukowiscydozy
SZPZOZ im. Dzieci Warszawy w Dziekanowie Leśnym
ul.M. Konopnickiej 65
05-092 Łomianki
Poradnia Mukowiscydozy
SZOZ nad Matką i Dzieckiem
ul. Polanki 119
80-308 Gdańsk
Klinika Pediatrii
Śląski Uniwersytet Medyczny Górnośląskiego Centrum Zdrowia Dziecka
ul. Medyków 16
40-752 Katowice
Wojewódzki Specjalistyczny Szpital Dziecięcy im. Świętego Ludwika
ul.Strzelecka 2
31-503 Kraków
Wojewódzki Szpital Specjalistyczny im. Mikołaja Kopernika
ul. Pabianicka 62
93-513 Łódź
I Katedra Pediatrii
Klinika Gastroenterologii Dziecięcej i Chorób Metabolicznych
ul. Szpitalna 27/33
60-572 Poznań
Klinika Pediatrii, Endokrynologii, Diabetologii, Chorób Metabolicznych i Kardiologii Wieku Rozwojowego
Samodzielny Szpital Kliniczny nr 1 im. Prof. Tadeusza Sokołowskiego PAM
ul. Unii Lubelskiej 1
71-252 Szczecin
Poradnia Alergologiczno-Pulmonologiczna
ul. Krasińskiego 29
50-450 Wrocław
W Polsce istnieje internetowa grupa wsparcia dla osób dotkniętych mukowiscydozą. Została powołana do życia przez Fundację Oddech Życia.
Po krótkie rejestracji dostępna jest możliwość rozmowy z zespołem Oddechu Życia oraz zarejestrowanymi innymi chorymi oraz ich rodzinami.
Na grupie można swobodnie porozmawiać o wszystkim co związane z mukowiscydozą i życiem. Na trudniejsze pytania odpowiadają nie tylko chorzy, rodzice, ale również pracownicy i wolontariusze Fundacji Oddech Życia.
Grupa wsparcia dla osób dotkniętych mukowiscydozą dostępna jest pod adresem https://www.facebook.com/groups/mukowiscydoza/