Dziedziczenie mukowiscydozy – podstawowe warianty
Jak dziedziczy się mukowiscydoza? Najważniejsze warianty przekazywania genu CFTR
Mukowiscydoza (cystic fibrosis, CF) jest jedną z najczęstszych chorób dziedzicznych w populacji europejskiej, w tym w Polsce. Choroba wynika z obecności zmian (wariantów) w genie CFTR, który znajduje się na chromosomie 7. Gen ten koduje białko CFTR – kanał jonowy odpowiedzialny przede wszystkim za transport chlorków i pośrednio za prawidłowe uwodnienie wydzielin w wielu narządach. Gdy funkcja CFTR jest istotnie zaburzona, wydzieliny stają się gęstsze i trudniejsze do usuwania, co sprzyja m.in. przewlekłym zakażeniom i zapaleniu dróg oddechowych, zaburzeniom trawienia i wchłaniania oraz wielu innym powikłaniom ogólnoustrojowym.
Mukowiscydoza dziedziczy się autosomalnie recesywnie, co oznacza, że aby rozwinęła się klasyczna postać choroby, dziecko musi odziedziczyć dwie patogenne (chorobotwórcze) zmiany w CFTR – po jednej od każdego z rodziców. Ponieważ gen CFTR leży na chromosomie autosomalnym, a nie na chromosomie płci, płeć dziecka nie wpływa na ryzyko dziedziczenia. W praktyce poradnictwa genetycznego ryzyko jest takie samo dla dziewczynek i chłopców.
W tym materiale opisujemy podstawowe warianty dziedziczenia, tak jak najczęściej przedstawia się je w edukacji pacjentów i rodzin. Jednocześnie dopowiadamy kilka kluczowych kwestii, które w krótkich opisach bywają pomijane, a mają znaczenie dla interpretacji wyników badań genetycznych i realnych decyzji rodzinnych.



Co dokładnie „dziedziczymy”: warianty genu CFTR i ich znaczenie
W języku potocznym często mówi się „mutacja CFTR”, jednak w medycynie i genetyce klinicznej coraz częściej używa się pojęcia „wariant” (zmiana w DNA), ponieważ nie każda zmiana w genie jest chorobotwórcza. W CFTR opisano bardzo wiele wariantów – część z nich jest jednoznacznie związana z mukowiscydozą (tzw. warianty CF-causing), część ma znaczenie niepewne (VUS), a część wiąże się raczej z łagodniejszymi lub narządowo ograniczonymi problemami z kręgu CFTR-RD (CFTR-related disorders) albo stanami granicznymi, jak CFSPID/CRMS rozpoznawane u części noworodków po badaniach przesiewowych.
Dlatego w poradnictwie genetycznym zawsze warto rozdzielić dwie rzeczy: matematykę dziedziczenia (kto może przekazać którą kopię genu) oraz interpretację kliniczną (co oznacza dana para wariantów u konkretnej osoby). Poniższe scenariusze procentowe są najbardziej przydatne wtedy, gdy mówimy o wariantach uznanych za patogenne i typowo wywołujących mukowiscydozę. Jeśli w grę wchodzą warianty o niepełnej penetracji, warianty o znaczeniu niepewnym albo sytuacje graniczne, ryzyko „choroby” może wymagać interpretacji przez genetyka klinicznego i zespół CF.
Jak rozumieć procenty ryzyka w kolejnych ciążach
W opisach dziedziczenia najczęściej podaje się ryzyko w procentach: 25%, 50%, 100% itd. Kluczowe jest, aby pamiętać o jednej zasadzie, która rodzinom bardzo często umyka w stresie: każda ciąża jest niezależnym „losowaniem”. To znaczy, że jeśli para ma np. 25% ryzyka urodzenia dziecka chorego, to nie działa to jak „licznik”, który po urodzeniu jednego zdrowego dziecka „musi” wyrównać statystykę w kolejnej ciąży. W praktyce może się zdarzyć, że w rodzinie z czwórką dzieci nie urodzi się ani jedno dziecko chore, ale może się też zdarzyć, że choroba wystąpi u więcej niż jednego dziecka – mimo że procenty „średnio” wyglądają inaczej.
Podstawowe warianty dziedziczenia mukowiscydozy
Dla przejrzystości przyjmujemy trzy proste określenia:
- rodzic bez patogennych wariantów CFTR (w uproszczeniu: „bez mutacji”),
- rodzic – bezobjawowy nosiciel jednej patogennej zmiany CFTR,
- rodzic chory na mukowiscydozę, czyli posiadający dwie patogenne zmiany CFTR.
W realnym świecie warto dodać jedno zastrzeżenie: „brak mutacji” w wyniku badań przesiewowych nie zawsze oznacza absolutny brak wariantów w CFTR, bo testy różnią się zakresem (panel najczęstszych wariantów, pełne sekwencjonowanie, analiza delecji/duplikacji). W scenariuszach poniżej chodzi jednak o logiczne modele dziedziczenia, które pomagają zrozumieć zasady.
1. Obydwoje rodzice bez zmutowanych genów CFTR.
To sytuacja, w której ani matka, ani ojciec nie mają patogennych zmian w CFTR. W takim układzie nie ma możliwości przekazania dziecku patogennego wariantu CFTR, a więc ryzyko urodzenia dziecka chorego na mukowiscydozę w klasycznym rozumieniu jest praktycznie równe 0%. Dzieci tej pary nie będą ani chore, ani nosicielami – jeśli rzeczywiście oboje rodzice nie posiadają patogennych wariantów.
W praktyce klinicznej zdarzenia typu „nowa mutacja de novo” w CFTR, prowadząca do mukowiscydozy u dziecka przy braku wariantów u rodziców, są uznawane za skrajnie rzadkie i zwykle pomijalne w standardowym poradnictwie. Jeśli jednak w rodzinie pojawiają się niejasne wyniki badań lub objawy sugerujące CF/CFTR-RD, interpretacja zawsze powinna być pogłębiona.

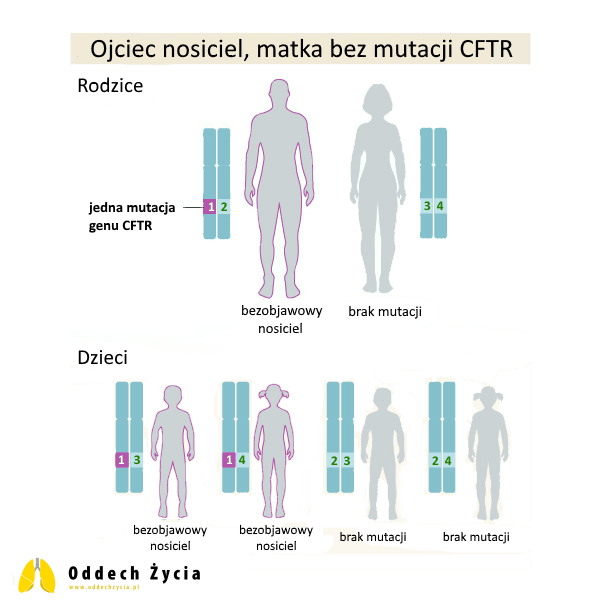
2. Ojciec nosiciel jednej mutacji CFTR, matka bez mutacji CFTR
Ojciec jest bezobjawowym nosicielem jednej patogennej zmiany CFTR. Nie choruje na mukowiscydozę, ponieważ ma jedną prawidłową kopię genu, która zwykle wystarcza, by nie rozwinęły się objawy choroby. Matka nie ma patogennych zmian w CFTR.
W tym układzie dla każdego dziecka:
- prawdopodobieństwo odziedziczenia patogennego wariantu CFTR od ojca wynosi 50%; takie dziecko będzie bezobjawowym nosicielem,
- prawdopodobieństwo odziedziczenia od ojca prawidłowej kopii CFTR wynosi 50%; takie dziecko nie będzie nosicielem,
- ryzyko mukowiscydozy wynosi 0%, ponieważ dziecko nie może otrzymać dwóch patogennych wariantów (matka nie ma czego przekazać w tym zakresie).
Warto tu dopowiedzieć rzecz bardzo praktyczną: w rodzinach często pojawia się zdanie „skoro ja jestem nosicielem, to dziecko na pewno będzie chore”. To nieprawda. Nosicielstwo samo w sobie nie oznacza choroby, a do rozwoju klasycznej mukowiscydozy potrzebne są dwie patogenne kopie genu – jedna od każdego z rodziców.
3. Matka nosicielka jednej mutacji CFTR, ojciec bez mutacji CFTR
To scenariusz lustrzany do punktu 2. Matka jest bezobjawową nosicielką, ojciec nie ma patogennych wariantów.
Dla każdego dziecka:
- 50% szans na urodzenie dziecka zdrowego, będącego bezobjawowym nosicielem,
- 50% szans na urodzenie dziecka zdrowego, nienosiciela,
- 0% ryzyka mukowiscydozy w klasycznym ujęciu.
W praktyce poradnictwa genetycznego podkreśla się, że nosicielstwo jest częste w populacji i zwykle ujawnia się dopiero „przy okazji” diagnostyki dziecka, badań przedkoncepcyjnych lub badań prenatalnych.

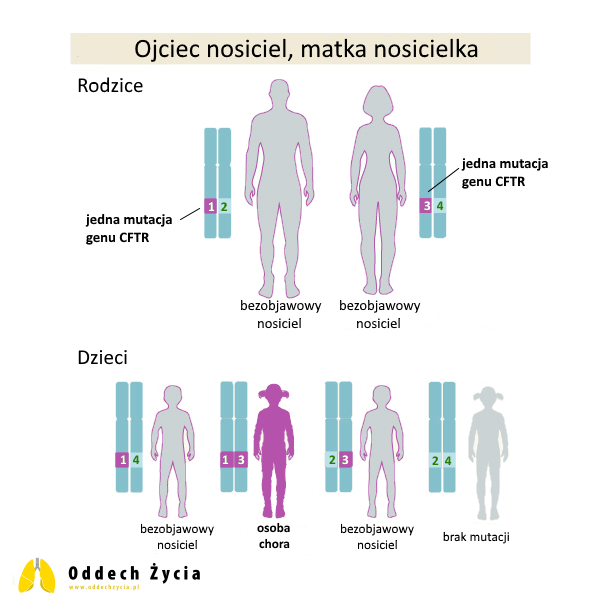
4. Ojciec i matka nosicielami genów CFTR
To najczęstsza sytuacja, w której rodzi się dziecko chore na mukowiscydozę: oboje rodzice są bezobjawowymi nosicielami po jednej patogennej zmianie w CFTR. W tym miejscu warto doprecyzować ważny szczegół: dla samej matematyki dziedziczenia nie ma znaczenia, czy rodzice mają tę samą zmianę, czy dwie różne patogenne zmiany. Natomiast dla przebiegu klinicznego choroby i możliwości leczenia przyczynowego (modulatorami CFTR) znaczenie konkretnego genotypu może być bardzo duże, co omówimy niżej.
Dla każdej ciąży w tym układzie istnieje:
- 25% szans na urodzenie dziecka bez mukowiscydozy i bez nosicielstwa (dziecko dziedziczy dwie prawidłowe kopie CFTR),
- 50% szans na urodzenie dziecka zdrowego, ale będącego nosicielem (dziecko dziedziczy jedną kopię prawidłową i jedną patogenną),
- 25% szans na urodzenie dziecka chorego na mukowiscydozę (dziecko dziedziczy dwie patogenne kopie – po jednej od każdego z rodziców).
To jest klasyczny schemat autosomalnego dziedziczenia recesywnego. Właśnie w tym miejscu najczęściej pojawiają się emocjonalne pytania: „dlaczego w naszej rodzinie wcześniej nie było mukowiscydozy?”. Odpowiedź jest prosta: nosicielstwo zwykle nie daje objawów, więc może być „ukryte” przez wiele pokoleń, aż do momentu, gdy dwoje nosicieli zostaje rodzicami.
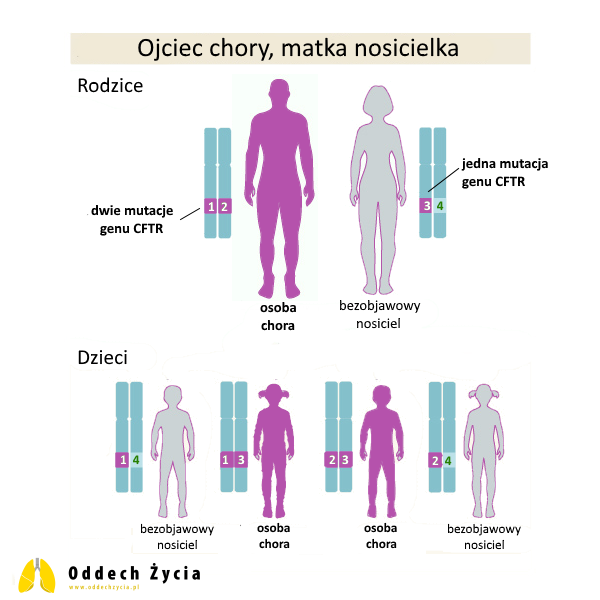
5. Ojciec chory na mukowiscydozę, matka bezobjawowa nosicielka jednej mutacji CFTR
W tej sytuacji ojciec ma dwie patogenne zmiany CFTR, a matka jedną patogenną i jedną prawidłową kopię CFTR.
Dla każdej ciąży:
- 50% szans na urodzenie dziecka chorego na mukowiscydozę (dziecko zawsze dostanie patogenną kopię od ojca i w połowie przypadków dostanie patogenną kopię od matki),
- 50% szans na urodzenie dziecka zdrowego, ale nosiciela (dziecko dostanie patogenną kopię od ojca i prawidłową kopię od matki),
- 0% szans na urodzenie dziecka bez żadnej patogennej kopii CFTR, ponieważ chory rodzic zawsze przekaże patogenną kopię genu.
To przykład, który dobrze pokazuje różnicę między pojęciami „zdrowy” i „bez żadnego ryzyka genetycznego”. Dziecko może być zdrowe (bez mukowiscydozy), ale w tym układzie nie może być całkowicie wolne od nosicielstwa, bo zawsze odziedziczy jedną patogenną kopię od chorego rodzica.
6. Matka chora na mukowiscydozę, ojciec bezobjawowy nosiciel jednej mutacji CFTR
To scenariusz analogiczny do punktu 5, tylko role płci są odwrócone. Matka ma dwie patogenne kopie CFTR, ojciec jest nosicielem.
Dla każdej ciąży:
- 50% szans na urodzenie dziecka chorego na mukowiscydozę,
- 50% szans na urodzenie dziecka zdrowego, ale nosiciela,
- 0% szans na urodzenie dziecka bez nosicielstwa.
Ponownie: płeć rodzica nie ma znaczenia dla matematyki dziedziczenia, bo CFTR nie jest genem sprzężonym z płcią.

7. Chorzy na mukowiscydozę rodzice
To sytuacja bardzo rzadka, ale w dobie lepszej opieki, modulatorów CFTR (leków przyczynowych) i wydłużenia życia osób z CF nie jest wyłącznie „teoretyczna”. Jeżeli oboje rodzice mają po dwie patogenne kopie genu CFTR, to każde dziecko odziedziczy patogenną kopię od matki i patogenną kopię od ojca.
W tym układzie:
- 100% dzieci będzie miało dwie patogenne kopie CFTR, czyli genetycznie będzie obciążone mukowiscydozą,
- 0% szans na dziecko bez choroby,
- 0% szans na dziecko będące jedynie nosicielem jednej patogennej kopii.
Warto jednak dodać istotne kliniczne dopowiedzenie: nawet przy „pewnym” dziedziczeniu dwóch patogennych kopii, fenotyp mukowiscydozy może się różnić między osobami (nawet w tej samej rodzinie), ponieważ na przebieg choroby wpływają m.in. inne geny modyfikujące, środowisko, mikrobiom, dostęp do leczenia i wczesność rozpoznania. Genetyka tłumaczy podstawę, ale nie opisuje całej historii choroby u konkretnej osoby.
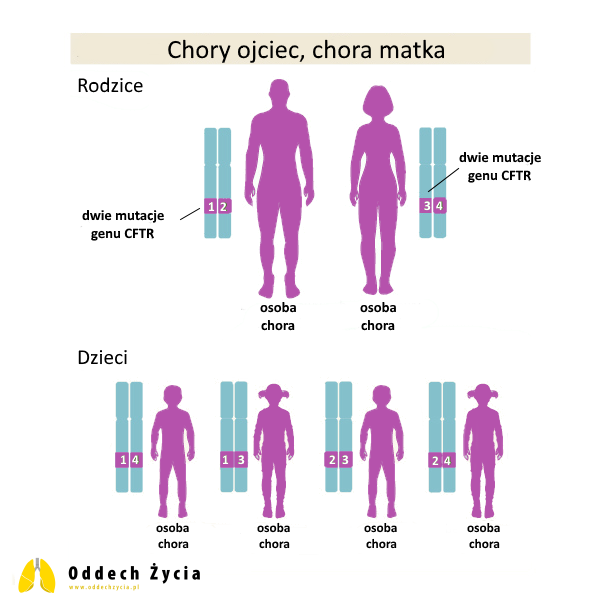
8. Chory na mukowiscydozę ojciec, matka bez mutacji CFTR
Ojciec ma dwie patogenne kopie CFTR, matka nie ma patogennych zmian.
Dla każdej ciąży:
- 100% szans na urodzenie dziecka zdrowego, ale będącego nosicielem jednej patogennej kopii CFTR,
- 0% szans na urodzenie dziecka bez nosicielstwa,
- 0% szans na urodzenie dziecka chorego na mukowiscydozę.
Ten scenariusz jest szczególnie ważny w edukacji rodzin, bo intuicyjnie wiele osób myśli: „skoro jedno z rodziców jest chore, to dziecko też musi być chore”. Tymczasem przy dziedziczeniu recesywnym tak się nie dzieje – jeśli drugi rodzic nie ma patogennego wariantu CFTR, dziecko nie uzyska dwóch patogennych kopii.
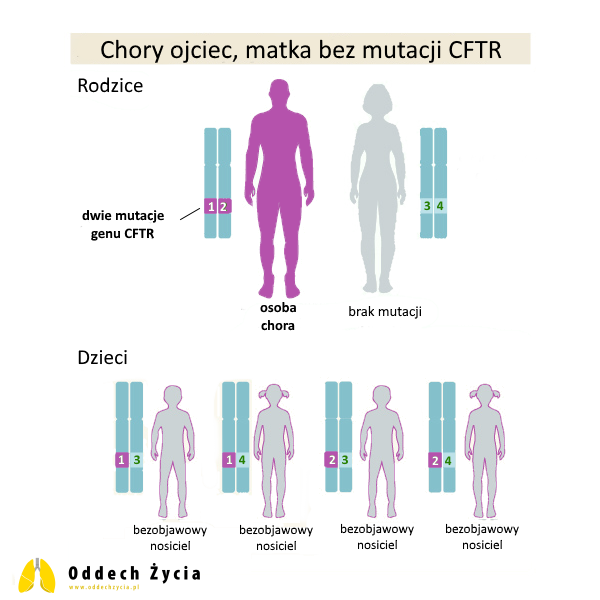
9. Chora na mukowiscydozę matka, ojciec bez mutacji CFTR
Sytuacja analogiczna do punktu 8: matka ma dwie patogenne kopie, ojciec nie ma patogennych wariantów.
Dla każdej ciąży:
- 100% szans na dziecko zdrowe, ale nosiciel,
- 0% szans na dziecko bez nosicielstwa,
- 0% szans na dziecko chore.

Dlaczego „taka sama czy różna mutacja” nie zmienia procentów, ale może zmieniać życie pacjenta
W edukacyjnych schematach często mówi się: „mogą to być takie same mutacje lub różne – nie ma to znaczenia”. Warto to zdanie doprecyzować, bo jest prawdziwe tylko w jednym sensie. Dla samych procentów dziedziczenia (czyli dla odpowiedzi na pytanie: „jakie jest ryzyko, że dziecko odziedziczy dwie patogenne kopie?”) zwykle nie ma znaczenia, czy rodzice są nosicielami tej samej zmiany, czy dwóch różnych patogennych zmian. Natomiast dla przebiegu klinicznego (np. ryzyka niewydolności zewnątrzwydzielniczej trzustki, tendencji do cięższych zakażeń, wartości chlorków w pocie, wieku rozpoznania) oraz dla kwalifikacji do leczenia przyczynowego (modulatorów CFTR) konkretna kombinacja wariantów może mieć bardzo duże znaczenie. Dlatego zawsze, gdy w rodzinie pojawia się wynik genetyczny, warto traktować go nie jako „etykietę”, ale jako informację kliniczną, którą powinien zinterpretować zespół zajmujący się mukowiscydozą.
Badania nosicielstwa i „ryzyko resztkowe”: dlaczego wynik ujemny nie zawsze oznacza „0%”
W praktyce pacjenci często pytają: „zrobiliśmy badanie i wyszło, że nie mamy mutacji – czy ryzyko znika?”. Odpowiedź zależy od tego, jakie badanie wykonano. Panele przesiewowe obejmujące najczęstsze warianty CFTR potrafią wykrywać dużą część nosicielstwa w populacji, ale nie obejmują wszystkich możliwych zmian. Ponadto nie każde badanie w standardzie wykrywa większe rearanżacje (np. delecje/duplikacje) czy rzadkie warianty w regionach trudniejszych analitycznie. Z tego powodu w genetyce mówi się o ryzyku resztkowym – czyli niewielkim, ale niezerowym ryzyku, które pozostaje mimo ujemnego wyniku ograniczonego panelu.
Z punktu widzenia rodziny najważniejsza jest praktyczna wskazówka: jeśli w rodzinie znany jest konkretny patogenny wariant CFTR (np. wykryty u dziecka), to badania u rodziców/partnera powinny być ukierunkowane tak, aby ten wariant z pewnością ocenić, a w razie potrzeby rozszerzyć diagnostykę. Takie decyzje podejmuje się najlepiej w ramach poradnictwa genetycznego, bo zakres badań powinien odpowiadać realnemu ryzyku i pytaniu klinicznemu.
Co zrobić, gdy w rodzinie jest mukowiscydoza albo planujecie ciążę
Jeśli w rodzinie występuje mukowiscydoza, jeśli macie dziecko chore lub jeśli jedno z Was jest nosicielem, najbardziej racjonalnym krokiem jest konsultacja w poradni genetycznej (często w porozumieniu z ośrodkiem prowadzącym mukowiscydozę). Celem takiej konsultacji nie jest „straszenie”, tylko uporządkowanie informacji: jaki wariant/warianty występują w rodzinie, jakie badania mają sens, jakie są realne ryzyka i jakie istnieją możliwości diagnostyczne na etapie planowania rodziny. W zależności od sytuacji rozważa się różne ścieżki, w tym diagnostykę przedkoncepcyjną (badania nosicielstwa u partnera), diagnostykę prenatalną w konkretnych wskazaniach lub – w części rodzin – diagnostykę preimplantacyjną w procedurach rozrodu wspomaganego. To są zawsze decyzje indywidualne, wymagające spokojnej rozmowy i dopasowania do wartości, zasobów i sytuacji medycznej danej pary.
Ważne jest też, aby pamiętać, że nawet w rodzinach bez rozpoznanej wcześniej mukowiscydozy nosicielstwo może być „niewidoczne” aż do momentu, gdy urodzi się chore dziecko. Z tego powodu w wielu krajach badania nosicielstwa oferuje się parom planującym ciążę, choć ich zakres i dostępność zależą od systemu ochrony zdrowia.
Mukowiscydoza i planowanie rodziny w erze modulatorów CFTR
W ostatnich latach opieka nad mukowiscydozą dynamicznie się zmienia, m.in. dzięki leczeniu przyczynowemu modulatorami CFTR u części pacjentów. Dla wielu dorosłych z CF oznacza to lepszą stabilność kliniczną, a w konsekwencji także częstsze rozważanie rodzicielstwa. Trzeba jednak pamiętać, że planowanie ciąży w mukowiscydozie powinno odbywać się w ścisłej współpracy z zespołem CF i położnikiem, ponieważ w grę wchodzą zagadnienia wydolności oddechowej, stanu odżywienia, ryzyka powikłań, farmakoterapii oraz monitorowania matki i dziecka. Dodatkowo w literaturze i zaleceniach zespołów eksperckich podkreśla się, że zwiększenie liczby ciąż obserwowane w praktyce może wiązać się m.in. z dostępnością ETI, a przy ekspozycji płodu/noworodka na modulatory mogą pojawiać się kwestie diagnostyczne, które wymagają czujności pediatrycznej i współpracy z ośrodkiem CF. Na poziomie rodziny najważniejsze jest jednak jedno: sama „era modulatorów” nie zmienia zasad genetyki – nadal obowiązuje to samo dziedziczenie autosomalne recesywne – ale może zmieniać kontekst kliniczny i organizacyjny opieki.
Zobacz też artykuły:
- Nowa mutacja de novo w CFTR – co to znaczy w praktyce
- Więcej niż dwie mutacje CFTR: allele złożone, cis-trans i rzadkie scenariusze genetyczne
- Dziedziczenie mukowiscydozy? Kto jest narażony? Jak przenosi się zmutowany gen?
Oddech Życia oraz U.S. National Library of Medicine


